
- den första godkända genterapin mot en ärftlig ögonsjukdom
Luxturna är ett bevis på att forskning och utveckling av innovativ precisionsmedicin ger resultat och innebär framsteg som vi för några år sedan inte vågade hoppas på. Luxturna är en genterapi som är avsedd för barn och vuxna som har synförlust förorsakad av hereditär retinaldystrofi på grund av bekräftade bialleliska RPE65 mutationer och som har tillräckligt med viabla näthinneceller.1
Sjukdomen orsakar tidigt i barndomen eller tonåren en betydande synnedsättning som förvärras med åren och leder till total blindhet. Det finns inga andra behandlingar än Luxturna med beprövad och visad effekt mot sjukdomen.
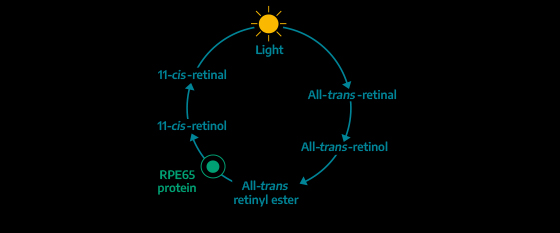
Luxturna innehåller en virusvektor som bär en fungerande RPE65-gen. När Luxturna injiceras subretinalt, levererar virusvektorn den fungerande genen till RPE-cellerna. Med en fungerande RPE65-gen kan RPE-cellerna producera normalt RPE65-protein så att syncykelns funktion återställs.1
Se filmen om Luxturnas verkningsmekanism
- Redan efter 30 dagar förbättras den funktionella synförmågan* signifikant med Luxturna, jämfört med kontrollgruppen.1,2
- Luxturna ger en >100-faldig förbättring av ljuskänslighet vid svagt ljus jämfört med kontrollgruppen.†1,2
- Signifikant förbättrat synfält med Luxturna, jämfört med kontrollgruppen.‡2
- Bibehållen synskärpa i 3 år visat i uppföljning av fas III-studien och i 4 år vid uppföljning av fas I-studien med Luxturna.§1,3
- Säkerhetsprofilen för Luxturna har studerats i tre kliniska studier med totalt 41 patienter. Det förekom retinala utfällningar hos 3 av 41 (7 %) personer och de upplöstes utan komplikationer.1
Studiens primära effektmått var den genomsnittliga förändringen mellan baslinjen och efter ett års behandling av patientens resultat i multiluminescensmobilitetstestning (MLMT) med båda ögonen.
För att kunna utvärdera den funktionella synförmågan hos patienterna före och efter behandling med Luxturna togs ett validerat binokulärt multiluminescensmobilitetstest (MLMT) fram. Med MLMT mäts förmågan hos en person att ta sig fram och navigera en rutt exakt och med rimlig hastighet vid olika nivåer av belysning i omgivningen. Denna förmåga beror på personens synskärpa, synfält och graden av nyktalopi (minskad förmåga att förnimma och/eller se i svagt ljus). Alla dessa funktioner påverkas av RPE65 mutationer.1
Fas III-studien var en öppen, randomiserad, kontrollerad studie. I studien deltog 31 patienter; 13 män och 18 kvinnor. Alla patienter hade diagnosen Lebers kongenitala amauros på grund av RPE65 mutationer som fastställts av ett certifierat laboratorium genom genetisk analys.
- 64 % var pediatriska patienter, n = 20, i åldrarna 4 år till 17 år
- 36 % var vuxna (n = 11)
Den genomsnittliga åldern för samtliga patienter var 15 år, från 4 år till 44 år.
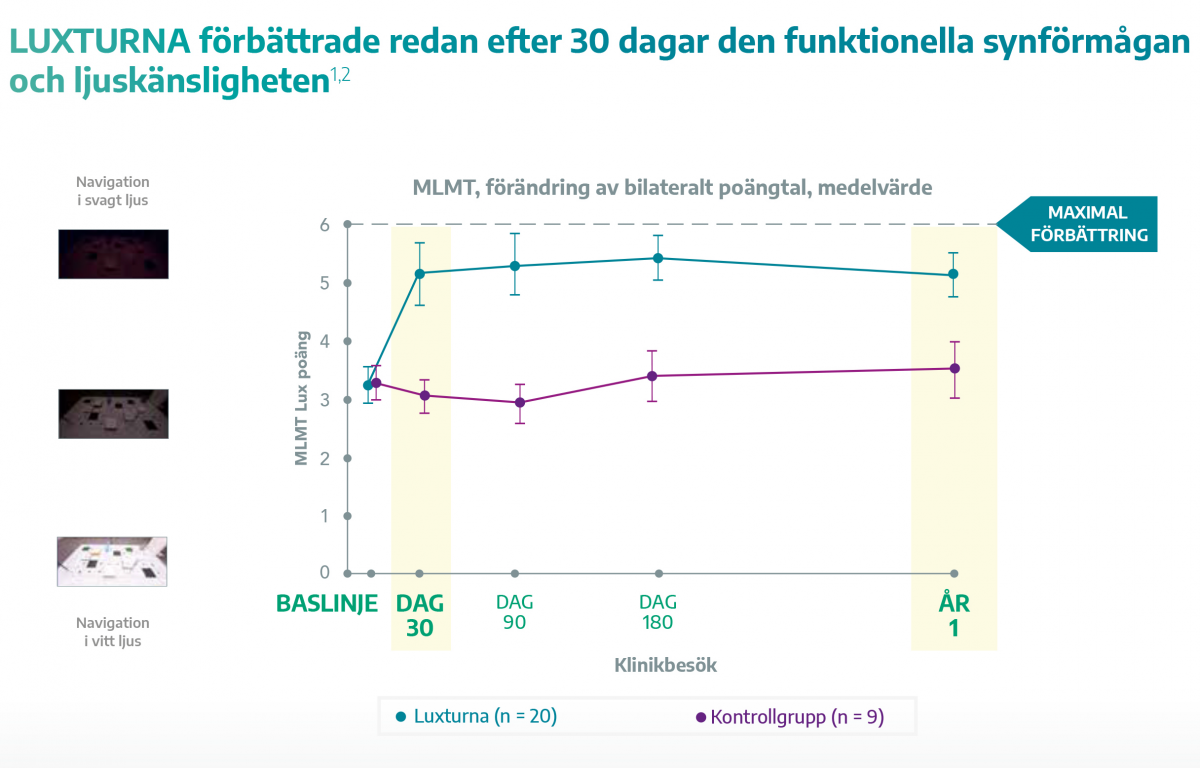
En ökning av poängtalet betyder att personen klarade MLMT på en lägre ljusnivå. Ett luxvärde på 6 betyder den bästa tänkbara förbättringen i MLMT. Vid varje test så videobandades personen och bedömdes av självständiga bedömare.1
Luxturna jämfört med kontroll, (95% KI, intervention-kontroll 1,6 (0,72 to 2,41) p= 0,00132.
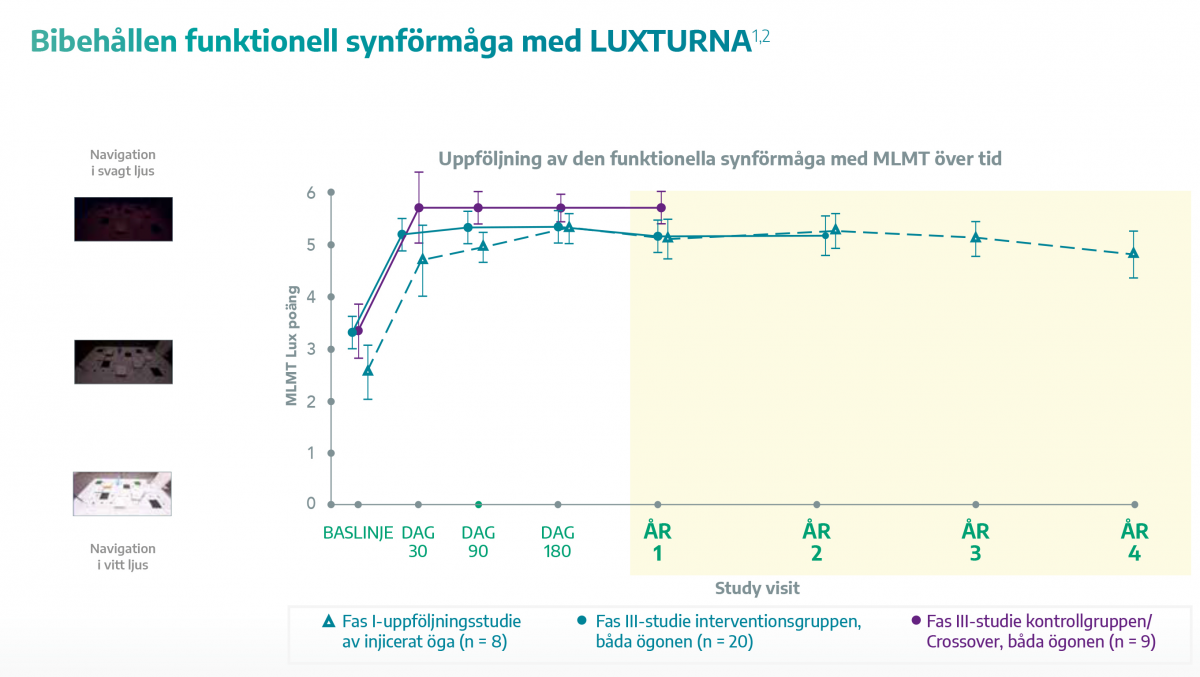
I fas III-studien genomfördes MLMT i sju nivåer av belysning från 400 lux till 1 lux, vilket motsvarar belysningen exempelvis från ett starkt belyst kontor till en månfri sommarnatt.1,3
FST mäter hur mycket ljusenergi som detekteras av ögat och speglar näthinnans ljuskänslighet. Metoden kan utföras även på patienter med dålig synskärpa, begränsat synfält och nystagmus.
Ett år efter behandling med Luxturna visade resultaten en ökning i ljuskänslighet >100 gånger jämfört med kontrollgruppen, (95% KI, intervention-kontroll): -2,33 (-3,44,-1,22,), p< 0,001.1 Förbättring i full-field light sensitivity (FST) har varit stabil upp till 4 år efter exponeringen för Luxturna.3
Två ytterligare sekundära effektmått testades: MLMT-resultat med monokulärt seende (för det först behandlade ögat, patientens sämsta öga vid baslinjen); och synskärpan (best-corrected visual acuity, BCVA) binokulär (båda ögonen). Även andra effektmått undersöktes i fas III-studien, exempelvis patienternas synfält.
I studien kan du ta del av ytterligare resultat:
Maguire AM, et al. Ophthalmology. 2019; 126(9): 1273–1285.
Långtidssäkerheten och -effekten av Luxturna bedömdes i tre kliniska studier med totalt 41 patienter. Det förekom tre icke-allvarliga biverkningar bestående av retinala utfällningar hos 3 av 41 (7 %) personer vilka ansågs vara relaterade till Luxturna. Alla dessa tre biverkningar bestod av övergående förekomst av symtomfria subretinala utfällningar nedanför det retinala injektionsstället, 1 6 dygn efter injektionen och de upplöstes utan komplikationer.
Under det kliniska utvecklingsprogrammet rapporterades allvarliga biverkningar förknippade med administreringsingreppet hos tre personer. 1 av 41 patienter rapporterade en allvarlig biverkning av ökat intraokulärt tryck (sekundärt till administrering av depåsteroid) som associerades med behandling av endoftalmit i association till administreringsförfarandet och ledde till optikusatrofi, och 1 av 41 patienter rapporterade en allvarlig händelse gällande näthinnestörning (förlust av foveal funktion) som bedömdes vara relaterad till administreringsförfarandet. 1 av 41 patienter rapporterade en allvarlig händelse gällande näthinneavlossning som bedömdes vara relaterad till administreringsförfarandet.
De vanligaste biverkningarna (incidens ≥ 5 %) som var relaterade till administreringsförfarandet var konjunktival hyperemi, katarakt, ökat intraokulärt tryck, näthinneperforation, dellen, makulahål, subretinala utfällningar, ögoninflammation, ögonirritation, ögonvärk och makulopati (veck på makulaytan).
* MLMT: Multiluminescensmobilitetstest; Primära effektmått fas III-studien. Luxturna jämfört med kontroll, (95% KI, intervention-kontroll 1,6 (0,72 to 2,41) p= 0,0011,2
† FST: full-field light sensitivity;Ett sekundärt effektmåttfas III-studien, FST vitt ljus [Log10(cd.s/m2)] för Luxturna visade resultaten en ökning i ljuskänslighet >100 gånger jämfört med kontrollgruppen, (95% KI, intervention-kontroll): -2,33 (-3,44,-1,22,), p< 0,001.1
‡ Goldmann III4e sum total degrees, genomsnitt av båda ögonen (monokulärt test), p=0.006; Humphrey macula threshold, genomsnitt av båda ögonen (monokulärt test); p=0.001) 2
§ En icke-signifikant trend av förbättrad synskärpa visades vid mätning med Holladay-skalan (p=0.27); När synskärpa utanför syntavlan beräknades med Lange-skalan visades en signifikant förbättring hos patienter jmf kontroll (p=0.0047).2
Referenser
-
Luxturna® SPC
-
Russell S, et al. Voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. The Lancet. 2017; 390(10097): 849-860.
-
Maguire AM, et al. Ophthalmology. 2019; 126(9): 1273–1285.